April 22, 2010
Dangerous plaques in blood vessels rupture by overproducing protein-busting enzymes
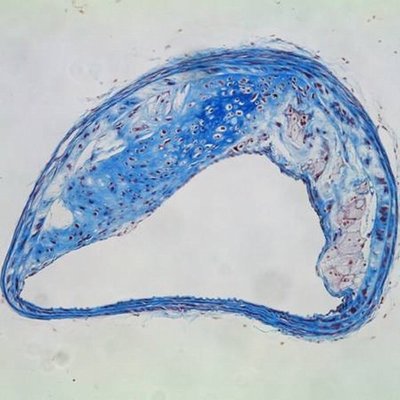
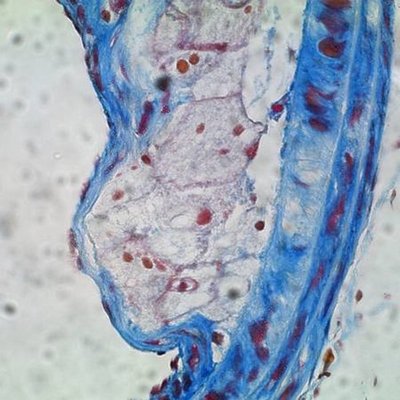
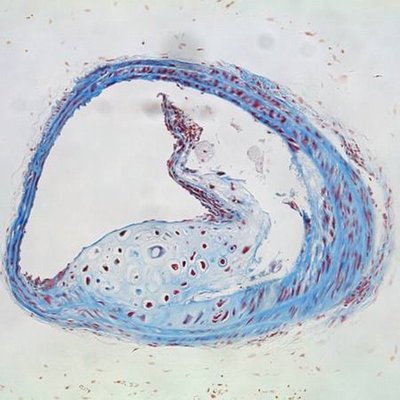
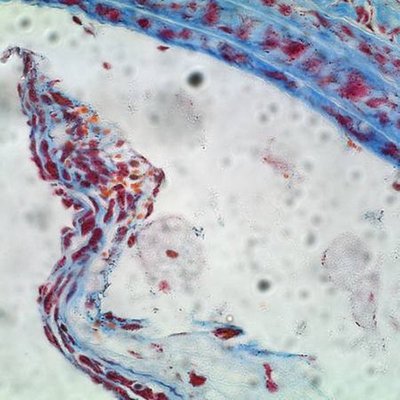
UW researchers have gathered evidence that dangerous plaques in blood vessels can rupture by overproducing protein-digesting enzymes.
Plaques are fat-laden rough spots in the otherwise smooth walls of arteries. When a plaque ruptures, blood accumulates inside of it, a process known as plaque hemorrhage. The plaque enlarges and artery-blocking clots can form. If the flow of oxygen-rich blood is restricted, a heart attack, stroke, or damage to other organs can occur.
Most adults have some degree of plaque in their arteries. The condition, known as atherosclerosis or “hardening of the arteries,” generally progresses with age. Not all plaques have a tendency to break open.
“Why plaques become unstable in certain people, and why some plaques become unstable but others plaques in the same person remain stable have been the objects of much conjecture,” said Dr. David A. Dichek, the senior researcher on the study, “Some scientists have suspected that an excessive production of protein-digesting enzymes might be linked to ‘vulnerable plaque’ — blood vessel lesions prone to rupture. The findings of our study offer the best cause and effect evidence to date that this is likely the case.”
The study was published online ahead of print March 29, in Circulation, a scientific journal of the American Heart Association.
Dichek holds the UW Medicine John Locke, Jr. Family Endowed Chair in Cardiovascular Research and Treatment and is a professor of medicine in the Division of Cardiology. He practices general cardiology at the UW Medicine Regional Heart Center.
People start life with clean arteries. Plaques appear when white blood cells burrow into the lining of arteries and engulf harmful cholesterol and lipids. Plaques begin as fatty streaks. Fatty streaks can attract several kinds of cells, including clean-up cells called macrophages. The plaques can enlarge until their cores are filled with dead cells, fats, and other debris. UW researchers learned that, inside some advanced plaques, macrophages churn out an overload of protein-digesting enzymes.
In a mouse study, Dichek and his team transplanted bone marrow containing macrophages that overproduce a protein-digesting enzyme known as urokinase into aged, atherosclerotic mice. These macrophages accumulated in some of the plaques. Compared to atherosclerotic mice of the same age, atherosclerotic mice with macrophages in overdrive had a much greater prevalence (61 percent versus 13 percent) of hemorrhage inside the plaques as well as more disruption of the fibrous caps covering the plaques.
“These same features — hemorrhage and cap disruption — are associated with ruptured plaques that cause heart attacks and strokes in humans,” Dichek noted.
Urokinase was already known to be abundant in ruptured human plaques.
“But automatically blaming urokinase as the culprit without experimental evidence that it destabilized otherwise stable plaques would have been like seeing bright red trucks and firefighters at every flaming building and assuming they had set the blazes,” Dichek said “In fact urokinase does have a protective effect at the right place and the right time, and we use it therapeutically as a clot-buster.”
The researchers also observed that the increase in urokinase upped the activity of other protein-digesting enzymes that can eat away at the structural proteins that hold a plaque together. These other enzymes can weaken the framework of the plaque and also promote the self-destruction of nearby smooth muscle cells in the artery.
These enzymes belong to a large family of remodeling enzymes called matrix metalloproteinases, or MMPs. The exact form of MMP implicated in this study hasn’t been determined. Like urokinase, MMPs have helpful roles, depending on the location and circumstances in the body.
Urokinase also activates a substance in blood vessels called plasminogen and turns it into plasmin, another protein-destroying enzyme. In several studies, patients whose blood tests showed a high level of plasminogen activation had an elevated risk of future heart attacks and strokes. The new mouse study, the researchers noted, helps explain this somewhat puzzling clinical finding. Their data suggests that high levels of urokinase in plaque, leading to increases in plasminogen activation, may be causally related to plaque rupture and major cardiovascular events.
An improved understanding of the underlying cellular and molecular mechanisms that cause plaques to rupture puts medical scientists in a better position to develop preventive approaches and more effective treatments for vulnerable plaque
“Our findings point to both diagnostic and therapeutic strategies,” Dichek noted. “We eventually may be able to identify patients who are more at risk for heart attacks and strokes than are others. At present this is very difficult to do simply by imaging plaques. We might find ways to prevent plaque rupture in these high- risk patients and thereby prevent many life-threatening or disabling cardiovascular events.”
Perhaps, he said, this would be through interventions that decrease production of protein-destroying enzymes in the artery wall or that block their activities. A caveat for this approach would be taking steps to avoid toxicity to the patient.
“On the other hand, if we discover that a specific MMP enzyme is found only this disease state, it would make an ideal treatment target,” Dichek added. Other options might be transferring genes into the blood vessel wall to lessen the manufacture of protein-digesting enzymes.
“But we have quite a long way to go before developing an effective gene transfer method and making it practical,” he said.
However, he added, “I hope it might eventually be possible to transform atherosclerosis into a condition that — like grey hair — is associated with aging, but which rarely (if ever) kills anyone. Atherosclerosis itself is a relatively low-risk condition: almost everyone over age 60 has it to some extent. Plaque rupture is what converts it from a chronic, indolent disease to an acute, life-threatening emergency. That is why plaque rupture is an important process to understand, track and control.”
In addition to Dichek, other scientists on this study, all from the UW, were Jie Hong Hu, acting instructor of medicine, Division of Cardiology; Liang Du, UW acting instructor of medicine; Talyn Chu, research scientist in cardiology; Goro Otsuka and Nagadhara Dronadula, both senior fellows in medicine and cardiology; Mia Jaffe, undergraduate student in biochemistry; Sean E. Gill, senior fellow in pulmonary and critical care medicine; and William C. Parks, professor of medicine in the Division of Pulmonary and Critical Care Medicine and director of the UW Center for Lung Biology.
The study was supported by a grant from the National Heart, Lung, and Blood Institute of the National Institutes of Health, a training grant for the student researcher from the Howard Hughes Medical Institute through the Undergraduate Science Education Program, and the John L. Locke, Jr. Charitable Trust.


