January 22, 2009
Study questions our understanding of early TB infection
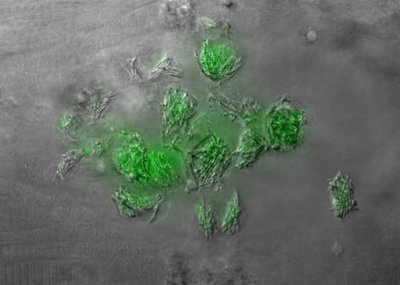
Tuberculosis bacteria can commandeer the body’s defenses in the early stages of infection and redirect them for their own offensive strategies, according to results reported Jan. 8 in the scientific journal Cell.
The dense granulomas characteristic of TB were originally thought to thwart infection by walling-off newly invading tuberculosis bacteria and preventing their spread. The new published findings suggest that these structures do just the opposite, at least in the early stages of infection.
“Tubercles or granulomas have long been considered host-protective. What we observed completely overturns the fundamental dogma on their role, at least early in infection,” said Dr. Lalita Ramakrishnan, UW associate professor of microbiology, medicine, and immunology, who conducted the study with Dr. J. Muse Davis of the Immunology and Molecular Pathogenesis Graduate Program at Emory University and a visiting scholar at the UW.
Granolomas are dense, complex clusters of immune cells, some of which are infected with TB bacteria. The outer edges of granulomas are surrounded by tightly interlocked white blood cells. Evidence from Ramakrishnan’s and Davis’ latest research suggests they might be created when newly arrived TB bacteria grow and emit signals to recruit macrophages — the body’s germ-eating and clean-up cells — to the initial infection site, then pirate the macrophages to truck fresh bacteria to other spots and replicate themselves there to establish more granulomas.
The granulomas form within days of infection. This occurs during the period when the body is trying to launch its first, generalized response to infection, called innate immunity. Later, when the body targets its fight, called adaptive immunity, Ramakrishnan says that the host and the bacteria usually reach a stalemate.
“One of the reasons we are studying the mechanisms of early TB infection,” Ramakrishnan said, “is to see where medical science might be able to intervene to stop the disease with new treatments without the problem of creating antibiotic resistance.” For example, she said, if scientists can figure out how TB bacteria signal to recruit and exploit macrophages and usurp the body’s immune processes to gain a foothold, perhaps researchers could then find a way to block this takeover.
By comparing fully virulent wild strains of bacteria that have a secretion system called ESX-1/RD1 with less virulent, mutant strains without this system, Ramakrishnan and Davis found that RD1 appears to encourage macrophages to arrive quickly at the site of infection. These macrophages moved five times faster than macrophages heading to the mutant bacteria, and covered greater areas with their haphazard movements.
The researchers saw that the fast macrophages took on the striking appearance of white cells racing to the scene after a chemical alarm: they had elongated centers, a ruffled front edge, and a trailing rear extension. The roundness of the slower macrophages going to the mutant bacteria indicated that they weren’t getting the same “hurry up” chemical signal.
Soon after arriving at the site of the wild-type, virulent bacteria, the macrophages became infected by engulfing the remains of other, previously infected macrophages, the researchers noted. RD1 is suspected to hasten the death of infected macrophages. The macrophages had collapsed like deflated balloons, but the waxy TB rods inside them were safe in the intact macrophage cell membrane. Several macrophages would help dispose of a single dead partner, thereby resulting in more infected cells for the bacteria to grow in. In contrast, the dying macrophages infected with mutant bacteria did not summon even nearby macrophages to come to them.
“This re-engulfing of virulent bacteria from dying macrophages is likely a major mechanism for granuloma expansion,” Ramakrishnan noted. The researchers observed that some of the newly infected macrophages left the original granuloma. The fluorescent-labeled bacteria inside them were later found at new granulomas elsewhere in the host.
“The bacteria in all the exiting macrophages remained fully viable after departure and had the potential to initiate new granulomas,” Ramakrishnan observed. “The macrophages exiting the original granuloma were leaving to seed distant granulomas to spread the infection.”
The role of RD1 in nascent granulomas revealed by this study, the researchers said, is likely a critical factor in the irrevocable early bacterial expansion that establishes the infection. Bacteria producing the RD1 virulence factor actually accelerate parts of the body’s innate immune response of macrophage recruitment, macrophage movement, and macrophage collapse and death, the researchers wrote. Instead of enhancing the body’s ability to halt a dangerous pathogen, the acceleration of these parts of the immune response is directed by a bacterial virulence factor and promotes bacterial growth, they concluded.
This study was supported by grants from the National Institutes of Health and the Burroughs Wellcome Foundation, as well as an American Society for Engineering Education predoctoral fellowship.


